While it's often useful to add a single carbon atom to a growing structure by using a carbene, it can sometimes be more convenient to add two carbon atoms at the same time. Such a dimer ( -C#C- )will usually bond readily to two radicals which are separated by an appropriate gap. If, for example, we remove two adjacent hydrogen atoms from the hydrogenated diamond (111) surface, we will have two radicals separated by ~0.25 nm. The use of a carbene to bridge a gap of this size is problematic, but a dimer should bond readily (Walch, 1996). Similar results for the edge of a graphite sheet are plausible.
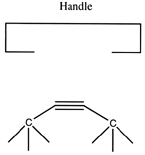
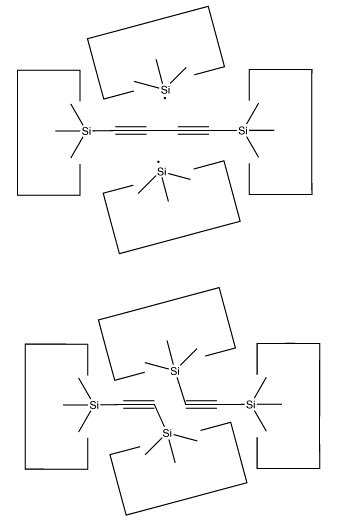
There are many possible dimer deposition tools -- the primary requirement is that the dimer be bonded relatively weakly to the rest of the tool so that after the dimer bonds to the desired target structure removal of the tool results in separation of the dimer from the tool. The general structure of a dimer deposition tool is illustrated below:
The sequence to deposit the dimer would then proceed as follows. First, the dimer would attach to the surface:
Reaction 16
Following this, the tool would be withdrawn:
Reaction 17
leaving the dimer on the surface.
A more flexible approach would be to form two separately controlled weak bonds to the dimer, as illustrated below.
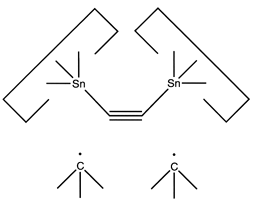
This allows the angle and strain of the C-Sn bond to be adjusted to suit the circumstances. The dimer would first be deposited on a suitably prepared site:
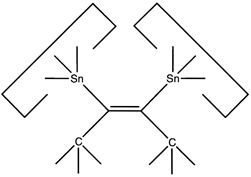
Reaction 18
After this, the first Sn moiety could be withdrawn:
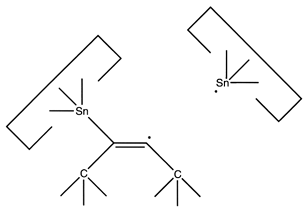
Reaction 19
and then the second:
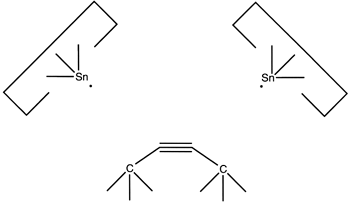
Reaction 20
resulting in deposition of the dimer on the surface, as desired.
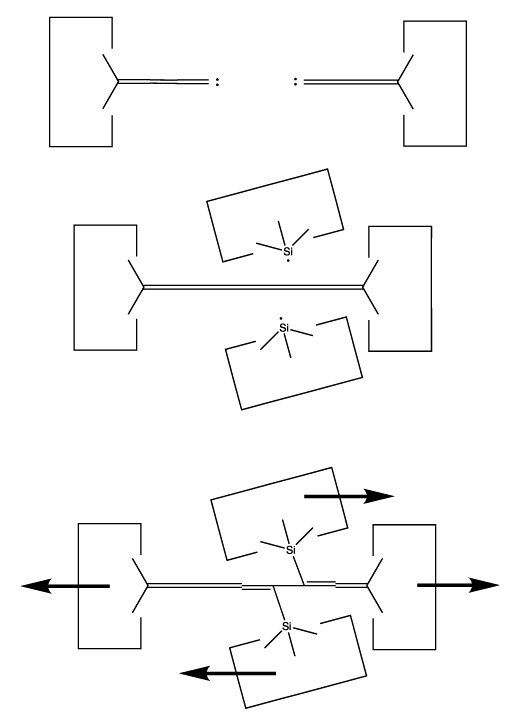
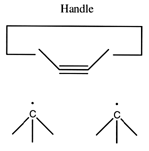
 A dimer deposition tool similar to a proposal by (Drexler, 1992), is illustrated at the left. This proposal has the useful property that separation of the dimer from its support causes a rearrangement of the bonding structure, thus eliminating any dangling bonds in the tool after the tool is withdrawn. However, it is possible that this dimer will rearrange to the undesired structure illustrated at the right. This issue needs to be investigated further.
A dimer deposition tool similar to a proposal by (Drexler, 1992), is illustrated at the left. This proposal has the useful property that separation of the dimer from its support causes a rearrangement of the bonding structure, thus eliminating any dangling bonds in the tool after the tool is withdrawn. However, it is possible that this dimer will rearrange to the undesired structure illustrated at the right. This issue needs to be investigated further.
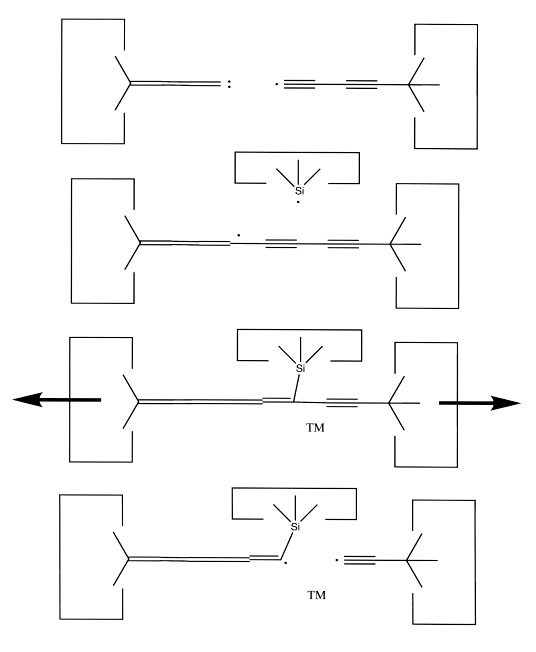
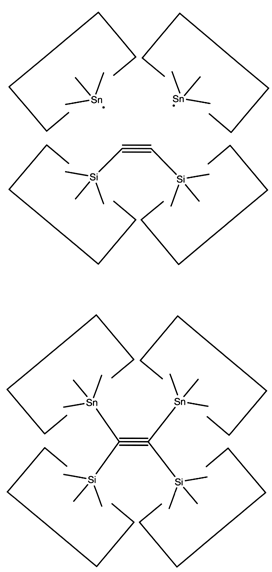
Once the dimer has been deposited on a surface and the tool withdrawn, the tool has been discharged and must be reloaded. Because dimer deposition tools are selected to bind weakly to the dimer (and hence to readily release the dimer when desired), reloading them is energetically unfavorable. In the following sequence we first split a four carbon polyyne chain (as provided by an earlier reaction) into two dimers, bonded at both ends to silicon. We then bend the Si-C#C-Si so that the reaction between it and the discharged dimer deposition tool is energetically favored. The two silicons are then rotated so that they are pointing at each other and force is applied until the approaching silicons form a Si-Si bond and break the Si-C bonds.
Reaction 21
Reaction 22
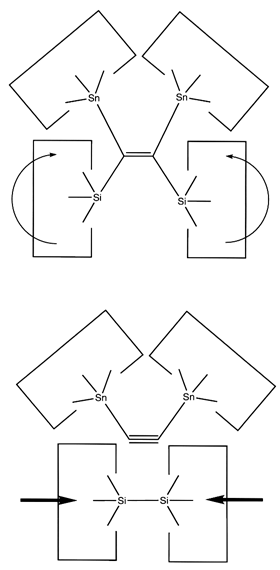
Reaction 23
Not all proposals will work
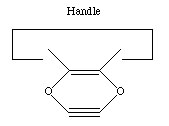
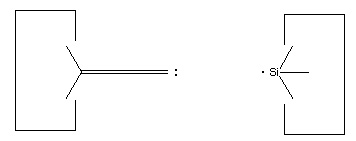
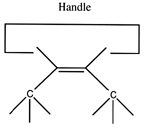
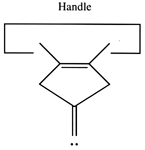
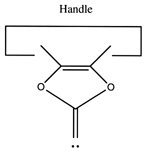 Another proposal for a dimer deposition tool is illustrated at the left. Ab initio calculations at the 6-31G* MP2 level using Gaussian (Frisch, 1995) show all positive vibrational frequencies for the dimer deposition tool, thus implying that it is stable in vacuum at a sufficiently low temperature. AM1 calculations show a barrier between this proposed dimer deposition tool and the isomeric carbene (illustrated at the right) of about 4 × 10-19 J (~60 kcal/mol). Unfortunately, more accurate (and computationally intensive) calculations at the 6-31G* Becke3LYP level show the barrier is only ~6 × 10-20 J (~8 kcal/mol. This does not include zero-point correction), almost an order of magnitude smaller. As a consequence, this dimer deposition tool is unlikely to work reliably at room temperature.
Another proposal for a dimer deposition tool is illustrated at the left. Ab initio calculations at the 6-31G* MP2 level using Gaussian (Frisch, 1995) show all positive vibrational frequencies for the dimer deposition tool, thus implying that it is stable in vacuum at a sufficiently low temperature. AM1 calculations show a barrier between this proposed dimer deposition tool and the isomeric carbene (illustrated at the right) of about 4 × 10-19 J (~60 kcal/mol). Unfortunately, more accurate (and computationally intensive) calculations at the 6-31G* Becke3LYP level show the barrier is only ~6 × 10-20 J (~8 kcal/mol. This does not include zero-point correction), almost an order of magnitude smaller. As a consequence, this dimer deposition tool is unlikely to work reliably at room temperature.
While not all proposals for molecular tools will work as desired, it is important to bear in mind that the computational methods used to evaluate such tools can be made increasingly accurate by increasing the computational effort devoted to the analysis. Our confidence in the conclusions can be increased by having more researchers analyze the problem with multiple methods and greater computational power.